RNAi for Zebrafish
- gutzman
- Jonathan Knight
UPDATED July 2012
Design and Use of shRNAs in Zebrafish
Jennifer H. Gutzman, Gianluca De Rienzo, and Hazel Sive*
Whitehead Institute for Biomedical Research and MIT
9 Cambridge Center
Cambridge, MA 02142
*present address
University of Wisconsin-Milwaukee
3209 N Maryland Ave.
Milwaukee, WI 53201
*See also De Rienzo, Gutzman and Sive, (July 2012, in press) Efficient shRNAmediated
inhibition of Gene Expression in Zebrafish. Zebrafish, Open Access.http://online.liebertpub.com/doi/abs/10.1089/zeb.2012.0770
Background:
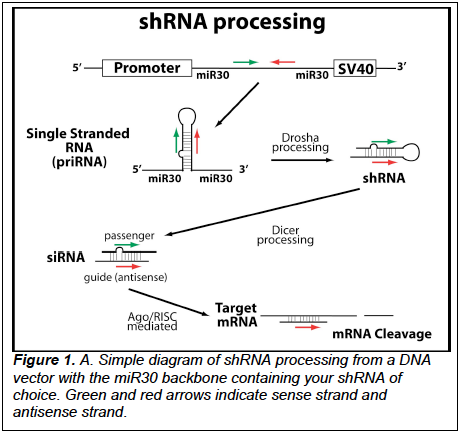
shRNAs (short hairpin RNAs) are processed into short interfering RNAs (siRNAs) that can
inhibit gene expression (Fig.1). Using a recently described procedure for zebrafish (Dong et al, 2009),
the miR30 gene transcribed region can be used to efficiently produce RNA hairpins, where the normal
miR30 hairpin is replaced with a hairpin targeting a gene of interest (GOI) (1). Hairpins are then
processed into interfering RNA. In general, it is most efficient to express the shRNA from a promoter.
Where the guide strand and target are exact matches, the target RNA is cleaved and subsequently
degraded by endogenous nucleases.
Proceed as follows.
I. Design a hairpin to your gene of interest (GOI). This will be part of a dsDNA molecule that will be
inserted into the miR30 backbone.
(see Detailed Protocols Section I: RNAi Hairpin Design)
II. Clone your GOI hairpin into the miR30 backbone. This step can be done either by
a. ordering two complementary, purified oligos, anneal these and subclone into the cut
vector. Such oligos run~$80 apiece, for a total of ~$160 per test hairpin.
b. ordering unpurified oligos, for ~$30 each (total ~$60) anneal and purify these yourself to save on cost
(see Detailed Protocols Section III: Oligo Purification/oligo annealing, phosphorylation and
ligation protocols below.)
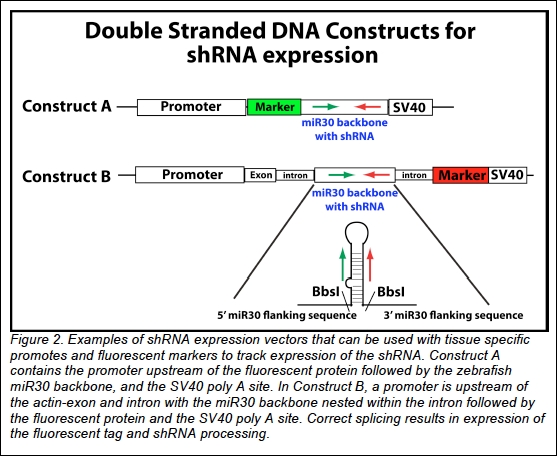
III. Place the miR30 backbone behind a ubiquitous or a tissue-specific promoter and a fluorescent
reporter, which can be transcribed from the same promoter. In Fig. 2 Construct A, contains the
promoter upstream of the fluorescent protein followed by the zebrafish miR30 backbone, and of the
SV40 poly A site. With this DNA construct, expression of GFP reflects transcription of the shRNA.
Alternatively, as in Fig. 2 Construct B, the miR30 backbone has been placed in the intron of the actinexon-
intron-fluorescent protein construct described by Dong et al. 2009 (1). After transcription, correct
splicing places the actin ATG in frame with the fluorescent tag; therefore, expression of the tag
indirectly indicates RNAi expression.
IV. Express the construct as a transgenic, usually initially, an F0 transgenic. Transgenesis can be
effected by meganuclease (I-SceI) or Tol2 depending on the vector backbone.
Monitor endogenous target RNA decrease using qPCR and in situ hybridization. Monitor phenotypes
and possible rescue by expression/injection of target RNA not subject to siRNA inhibition. (see
protocols section IV.)
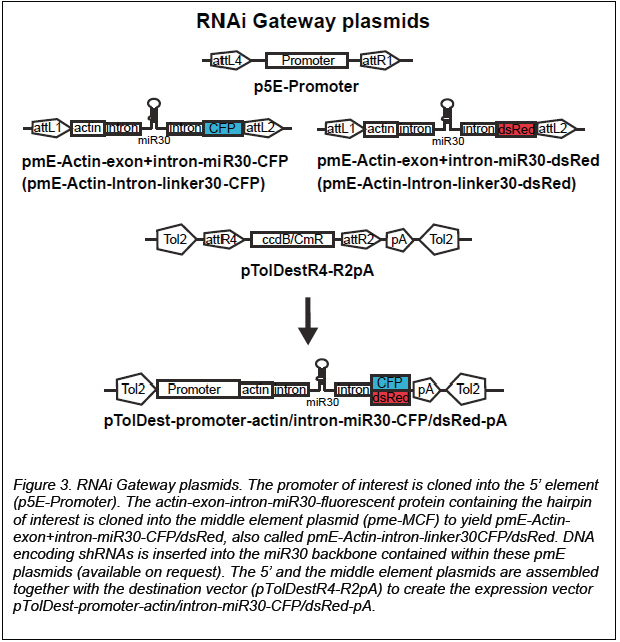
An easy way to allow interchanging of promoters and is through use of the Gateway system (Fig. 3)
(2,3). Transgenesis is generally obtained by Tol2 methods.
DETAILED PROTOCOLS
I. RNAi hairpin Design
The protocol below describes design and cloning of oligos containing a hairpin that will be inserted
into a miR30 backbone.
1. Use the Invitrogen Block-IT RNAi designer website for finding shRNA sequences that would target
your gene of interest.
2. Use this link and choose to design an shRNA, then enter your sequence or accession number.https://rnaidesigner.invitrogen.com/rnaiexpress/setOption.do?designOption=shrna&pid=- (https://rnaidesigner.invitrogen.com/rnaiexpress/setOption.do?designOption=shrna&pid=\-)
248931025050769267
3. Choose hits that are 4-5 stars and blast the sequences to the zebrafish genome to be sure your
target gene is your only hit (anything else should be looked at more carefully since off target effects
are possible depending on your target sequence and region of genome match within the hairpin).
4. Your shRNA target sequence should be a 21 mer.
5. Design 4 – 6 shRNAs per target. These can be in the coding sequence, or 3’UTR (De Rienzo et al,
2012).
6. Follow the example below to add in your sequence for designing your RNAi oligo. (should be a 71
mer).
7. YFP shRNA design example
Enhanced YFP NCBI ACCESSION: DI026212
1 atggtgagca agggcgagga gctgttcacc ggggtggtgc ccatcctggt cgagctggac
61 ggcgacgtaa acggccacaa gttcagcgtg tccggcgagg gcgagggcga tgccacctac
121 ggcaagctga ccctgaagtt catctgcacc accggcaagc tgcccgtgcc ctggcccacc
181 ctcgtgacta ccttcggcta cggcctgatg tgcttcgccc gctaccccga ccacatgaag
241 cagcacgact tcttcaagtc cgccatgccc gaaggctacg tccaggagcg caccatcttc
301 ttcaaggacg acggcaacta caagacccgc gccgaggtga agttcgaggg cgacaccctg
361 gtgaaccgca tcgagctgaa gggcatcgac ttcaaggagg acggcaacat cctggggcac
421 aagctggagt acaactacgg tggatccggt gctagcaaca gccacaacgt ctatatcatg
481 gccgacaagc agaagaacgg catcaaggtg aacttcaaga tccgccacaa catcgaggac
541 ggcagcgtgc agctcgccga ccactaccag cagaacaccc ccatcggcga cggccccgtg
601 ctgctgcccg acaaccacta cctgagctac cagtccgccc tgagcaaaga ccccaacgag
661 aagcgcgatc acatggtcct gctggagttc gtgaccgccg ccgggatcac tctcggcatg
721 gacgagctgt acaagtaa
pink = 21 mer Invitrogen Block-IT design 5 stars (THIS HAS NO BLAST HITS IN THE ZEBRAFISH
GENOME)
8. Oligo design with color coding (YFP example, continued)
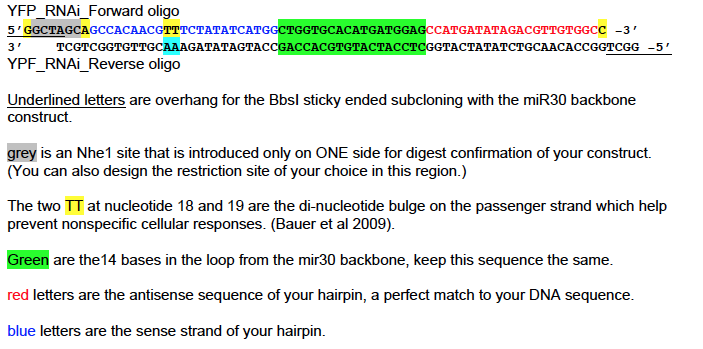
**NOTES:
All parts of the construct stay the same for each new targeting sequence EXCEPT FOR THE
RED AND BLUE LETTERS. Be sure to properly create your 3’ to 5’ compliment.
When ordering, remember that the two oligos above, that you anneal, are slightly different
because of the NheI restriction site only on one side so you cannot have the program do an
automatic reverse compliment.
II. Oligo Urea-PAGE Purification Protocol
1. Pour a large (~6inch X 6inch) denaturing UREA – PAGE gel.
% of your gel depends on the oligo size.
70 mer ∼ 8-10%
using National diagnostics reagents.
No. EL-830
835 {order EL-833 (kit with all three)
840
The gel thickness depends on the amount you need to purify.
You will get a band at the correct size – then a smear of unpure oligo below your correct size
band. For single nucleotide resolution decrease your starting material and run the gel longer.
2. Resuspend lyophilized oligo in H2O
Add 100 μl H2O to 50nmol of oligo and resuspend oligo by mixing at room temperature.
take about 50-20 μl oligo in H2O
add 2x loading dye
* Use loading dye containing both bromophenol blue and xylene cyanol with 8 M urea.
RECIPES
Urea Gel Loading Dye
2X mix is:
8M Urea (FW 60.06)
25mM EDTA (FW 372.2)
Use:
Urea: RSB gold capped molecular biology grade (cat. US 75826 1kg)
EDTA: Sigma molecular biology grade (cat. E-5134 500g)
Make:
1000mL, RNase-free (use no glass containers). Use only weigh boats from middle of stack
and only plastic containers from Corning 0.22um filter apparatus. You will need 2 500mL filter
apparatuses and 2 1000mL apparatuses (one to mix and one to actually filter).
Prepare 500mL, 500mM EDTA stock at pH 8.0 (see Maniatis 3 B.11)
a. to 93.1g EDTA, add 10g NaOH pellets; dump into 500mL filter bottle.
b. add nuclease-free H2O to 500mL
c. mix on orbital shaker until dissolved (overnight)
d. filter
Dissolve urea:
a. add 480g urea to 1000mL filter bottle
b. add nuclease-free H2O to 900mL
c. mix on orbital shaker until dissolved (overnight)
d. add 50mL, 500mM EDTA
e. top-off to 1000mL with nuclease-free H2O
f. filter
Preparing dyes to aliquot: we use 0.025% Xylene Cyanol and Bromophenol Blue, so weigh
out 0.0125g per 50mL.
3. Load/Run gel with 0.5 x TBE running buffer
Start @ 2 watts 10-15 mins
Increase to 10-15 watts
Check the xylene dye front to determine how far to run your gel
Xylene runs at 55 bp with 8% gel
4. When finished take gel apart and put the gel between 2 pieces of saran wrap.
5. Look at the gel with UV shadowing over fluor-coated TLC plate.
Plates available from Applied Biosystems AM10110 10x10cm ~$50
Put saran wrapped gel on the plate – Quickly scan with UV light – Look for shadows. (The
shadows are your bands and indicate the presence of DNA blocking the UV light)
Draw on the saran wrap around Shadow with Sharpie (do Not cut your gel on the Plate)
6. Cut out gel pieces and put the gel containing your oligo in an eppendorf tube.
Cut into pieces if necessary –
Want gel piece about ~1square centimeter (too big will lead to impure oligo!) You do not want
to include any of the smear below your pure large band.
7. Add 400 μl 0.3M NaCl in H2O to tube (400 μl/tube of gel).
Rotate overnight @ 4°C.
DNA will come out of gel into salt!
8. Spin tube – acrylamide piece will be at bottom.
Collect Supernatant = oligo.
9. Add 2.5 x volumes of ethanol
2.5 x volumes 400ul. (so 1 ml for 400 μl)
into -20°C at least 2 hrs.
10. Spin on high --
oligo= pellet! (10 min @4°C)
Dry pellet!
Resuspend in 50μl nuclease free water and spec for your actual DNA oligo concentration
11. Use for annealing / oligo phosphorylation/ ligations (see protocols below).
**NOTES:
Should recover ∼ 25% of amount of oligo loaded on gel.
For example, based on starting material of ~ 660 μg loaded
Predict with 25% recovery = ~ 150 μg
Add 50 μl nuclease free water to each tube
Do spec reading
If see a range of 900 ng/μl – 2500 ng/μl, this means good recovery.
III. Oligo annealing, phosphorylation and ligation protocol
Following oligo purification, oligos are annealed, checked, phosphorylated and used as insert for
ligation your specific promoter driven vector of choice with the miR30 backbone and a fluorescent tag.
Annealing
Following oligo purification, oligos are annealed in a 50μl reaction
1μl of 100μM forward oligo
1μl of 100μM reverse oligo
25μl of annealing buffer
23μl of water
TOTAL 50μl
Annealing buffer: 200mM postassium acetate, 60mM HEPES-KOH pH 7.4, 4mM magnesium
acetate
(Another option for re-suspension of purified oligos and use as an annealing buffer:
10 mM Tris, pH 7.5--8.0, 50 mM NaCl, 1 mM EDTA)
For the annealing reaction:
Use a PCR machine, run the following program:
4’ 95°C
10’ 70°C
2’ 60°C
2’ 50°C
2’ 40°C
2’ 30°C
2’ 20°C
2’ 10°C
store at 4°C before use
Check for oligo annealing by running your annealed oligos, compared to single oligos, on a 2% DNA
agarose gel. The annealed oligos will run slower than the singles.
Oligo Phosphorylation
Annealed oligos 400 ng
10X ligation buffer (supplied with 10mM ATP) 3 μl
10 U/μl T4 polynucleotide kinase 2 μl
H20 to 30 μl
Incubate at 37°C for 30 minutes. Use directly for the ligation reaction.
Final concentration of the phosphorylated annealed oligo pair: ~13 ng/μl
Ligation reaction
Combine vector (containing miR30 backbone with BbsI sites) and the insert (annealed oligo) at a
molar ratio of 1:5
Prior to ligation, de-phosphorylate your vector using Antarctic Phosphatase treatment (New England
Biolabs).
Ligation reaction example:
vector (length 4479) 100 ng
insert (length 68) 7.6 ng
10X ligase buffer 2 μl
400U/μl ligase 1 μl
Water X μl
TOTAL 20μl reaction
Incubate
12 h at 15°C
Transformation
Transform DH5α cells with 5 μl of the ligation reaction
30’ on ice
5’ at 42°C
5’ on ice
add 300 μl of SOC media
1 h at 37°C
Plate with appropriate antibiotic
IV. Use of shRNAs in transient transgenic assays
Transgenic preparation
Initial analysis can be performed in transient (F0) transgenics.
For Tol2 vectors:
Combine:
125 ng/μl transgenic plasmid 4 μl
125 ng/μl Tol2 mRNA 4 μl
H20 12 μl
Inject 1nl of the solution into one-cell-stage eggs.
The mass amount of plasmid and transposase injected per embryo will be 25 pg of each.
Note
We found that this concentration is the best ratio for most of the constructs giving high fluorescence
and no necrosis. To increase fluorescence, it is possible to titrate the DNA up to 70 pg but it will
increase necrosis. To avoid necrosis it is possible to coinject 3 ng of p53MO (4).
Expect between 20-50% of dead/deformed embryos as a sign that the transposase is active and
transposition is occurring.
For meganuclease vectors:
Combine
150 ng/μl transgenic plasmid 4 μl
5 U/μl ISce-I 2 μl
10X ISce-I buffer 2 μl
H20 12 μl
Inject 1nl of the solution into one-cell-stage eggs
The concentration of plasmid/meganuclease injected per embryo will be 30 pg/0.5 U
Note
We found that this concentration is the best ratio for most of the constructs giving high fluorescence
and no necrosis. To increase fluorescence it is possible to titrate the DNA up to 70 pg but it will
increase necrosis.
Subsequent steps
• Score embryos for fluorescent protein expression.
• Quantify gene knockdown by qPCR.
• Score fluorescent embryos for phenotype.
• Attempt rescue of phenotype by injection or expression of target gene RNA that does not bind
siRNA.
REFERENCES
1. Dong M, Fu YF, Du TT, Jing CB, Fu CT, Chen Y, Jin Y, Deng M, Liu TX. Heritable and lineagespecific
gene knockdown in zebrafish embryo. PloS one. 2009;4, e6125.
2. De Rienzo G, Gutzman J, Sive H. Efficient shRNA-mediated inhibition of Gene Expression in
Zebrafish. Zebrafish. July 2012. http://online.liebertpub.com/doi/abs/10.1089/zeb.2012.0770
3. Villefranc JA, Amigo J, Lawson ND. Gateway compatible vectors for analysis of gene function in
the zebrafish. Dev Dyn. 2007;236:3077-3087.
4. Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ,
Kanki JP, Chien CB. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon
transgenesis constructs. Dev Dyn. 2007;236:3088-3099.
5. Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, Ekker SC. p53 activation
by knockdown technologies. PLoS Genet. 2007;3:e78.